Síndrome de Marfan
Revisado por pares por Dr Rosalyn Adleman, MRCGPÚltima actualización por Dr Philippa Vincent, MRCGPLast updated 3 Mar 2025
Cumple con las directrices editoriales
- DescargarDescargar
- Compartir
- Language
- Discusión
- Versión en audio
- Add to preferred sources on Google
En esta serie:Aneurisma aórtico abdominalDisección aórtica
El síndrome de Marfan (nombrado así por el Dr. Antoine Marfan, el médico francés que lo describió por primera vez en 1896) es un trastorno genético con el que las personas nacen y que dura toda la vida.
El síndrome de Marfan puede afectar a diferentes personas de diferentes maneras. Algunas personas tienen síntomas leves; otras personas tienen síntomas más severos.
Es posible que tengas antecedentes de síndrome de Marfan en tu familia. Si tienes síndrome de Marfan, tienes un 50% de probabilidad de transmitir la condición a cada uno de tus hijos.
At a glance
Marfan syndrome is a rare genetic condition affecting connective tissue in the body.
It can affect the heart, blood vessels, eyes, and skeleton.
Symptoms vary widely, from mild to severe, and often worsen with age.
Heart problems are the most serious and require regular monitoring.
Treatment can help manage problems caused by the syndrome.
Seek immediate medical help for sudden, severe chest pain, eye changes, or breathing difficulties.
En este artículo:
Video picks for Condiciones genéticas
Continúa leyendo abajo
¿Qué es el síndrome de Marfan?
Si tienes el síndrome de Marfan, tienes un problema con el tejido conectivo en tu cuerpo. (Tu tejido conectivo es el tejido que une, protege y da fuerza a otros tejidos y órganos dentro de tu cuerpo. También ayuda a mover grasa y nutrientes por tu cuerpo y puede ayudar a reparar daños en otros tejidos.) En el síndrome de Marfan, tu tejido conectivo se vuelve débil y 'flácido'.
Marfan syndrome can affect the connective tissue in different parts of your body, including your heart, blood vessels, eyes and skeleton.
Marfan syndrome is rare and is thought to affect about 1 in 3,000 to 1 in 5,000 people. It affects males and females equally. It affects people of all ethnicities equally.
It is the most common genetic problem affecting connective tissue. It is an autosomal dominant condition, meaning you only need one parent, not both, to have it to inherit it yourself.
También puede surgir "de novo", lo que significa que el gen que causa el síndrome de Marfan no ha sido heredado, sino que ha "mutado" (cambiado) durante los primeros desarrollos después de la concepción. Este es el caso en 1 de cada 4 personas con síndrome de Marfan, quienes, por lo tanto, no tendrán un padre afectado.
Síntomas del síndrome de Marfan
Volver al contenidoEl síndrome de Marfan puede afectar diferentes partes de tu cuerpo. No todas las partes del cuerpo están afectadas en todas las personas. Algunas personas con síndrome de Marfan pueden tener solo unos pocos síntomas o problemas, mientras que otras están más gravemente afectadas. Los síntomas tienden a empeorar a medida que envejeces.
La siguiente lista enumera los problemas y síntomas más comunes que pueden desarrollarse. Hay tratamientos disponibles para la mayoría de estos problemas. Los tratamientos se enumeran más adelante en el artículo.
El corazón y los vasos sanguíneos
See also the separate leaflet Anatomía del corazón.
Los problemas del corazón y los vasos sanguíneos son los problemas más graves que tienden a tener las personas con el síndrome de Marfan.
Marfan syndrome can weaken the wall of the main artery in your body (the aorta). Arteries carry blood from your heart to the rest of your body. The aorta runs from your heart, through the centre of your chest and then through the centre of your abdomen. Because the walls of the aorta become weak, it can widen or bulge out. This is known as an aortic aneurysm. Consulte el folleto separado llamado Aneurisma de la aorta abdominal para más detalles.
Aproximadamente 8 de cada 10 personas con síndrome de Marfan tienen ensanchamiento de esta arteria principal. En casos graves, la arteria ensanchada puede desgarrarse (consulte el folleto separado llamado Disección aórtica para más detalles) o ruptura. Esto puede causar sangrado severo y muerte. La dilatación de la arteria principal puede diagnosticarse a una edad temprana y luego puede empeorar con el tiempo.
Algunas personas con síndrome de Marfan tienen un problema con una de las válvulas de su corazón. Las válvulas separan las cámaras del corazón. El problema se llama prolapso de la válvula mitral y ocurre porque la válvula llamada válvula mitral se vuelve flácida. Su médico puede escuchar un soplo cardíaco cuando escucha su corazón. See the separate leaflet called Mitral regurgitation for more details.
El esqueleto
Las personas con síndrome de Marfan suelen ser más altas y delgadas que otros miembros de su familia que no tienen el síndrome de Marfan.
Tus brazos y piernas pueden ser más largos que los de otras personas.
Tus dedos pueden ser largos y delgados. Esto se llama aracnodactilia.
Su esternón puede estar presionado hacia adentro y sus costillas se curvarán hacia adentro con él. Esto se llama pecho hundido (pectus excavatum). Esto puede empeorar con el tiempo y causar dolor en el pecho y problemas respiratorios.
A veces el esternón se empuja hacia afuera, de modo que el pecho es más redondeado. Esto se llama pecho de paloma (pectus carinatum). Generalmente es indoloro y no causa problemas de salud.
More than 6 out of 10 people with Marfan syndrome have a sideways curve of the backbone (scoliosis). It can cause back pain. In severe cases it can put pressure on the heart and lungs and cause breathing problems. Consulte el folleto separado titulado Escoliosis y cifosis (Curvatura de la columna vertebral) para más detalles.
Los pies planos también son comunes.
Puede haber problemas con las articulaciones de la cadera, lo que lleva a dolor de cadera, rigidez y problemas para caminar. Esto eventualmente puede causar artritis en las articulaciones de la cadera.
Es posible que tengas articulaciones que estén flojas y más flexibles de lo habitual.
Los problemas dentales y bucales pueden incluir un paladar alto y arqueado, una mandíbula inferior pequeña y apiñamiento de los dientes.
Los ojos
En aproximadamente la mitad de las personas con síndrome de Marfan, el cristalino del ojo se desplaza a una posición anormal (llamada luxación del cristalino). Puede estar presente al nacer o desarrollarse cuando eres niño o adolescente.
Part of the lining of your eye (the retina) may tear or peel away from the back of your eye. This is called a retinal detachment. Consulte el folleto separado llamado Desprendimiento de retina para más detalles.
Otros posibles problemas oculares incluyen la miopía, la opacidad del cristalino dentro de su ojo (una catarata) y un aumento de la presión dentro de su ojo (glaucoma).
Los problemas oculares en personas con síndrome de Marfan pueden causar problemas de visión si no se tratan lo suficientemente temprano.
Otras características
An air leak from one of your lungs may make the lung collapse. This is called a spontaneous pneumothorax. It will cause sudden chest pain and make you short of breath. Consulte el folleto separado llamado Neumotórax para más detalles.
También puede notar estrías si tiene el síndrome de Marfan. Las estrías son líneas rojas/rosadas o blancas en su piel. Son comunes durante el embarazo o en personas que han ganado o perdido mucho peso o han crecido rápidamente. En personas con el síndrome de Marfan, debido a que la piel es más débil de lo normal, las estrías ocurren más fácilmente. Por lo general, se encuentran en los hombros, la espalda y los muslos.
Tu cerebro y médula espinal están rodeados por una membrana especial llamada duramadre. En personas con síndrome de Marfan, la duramadre puede debilitarse y expandirse hacia afuera. Esto se llama ectasia dural, que ocurre con mayor frecuencia en la zona lumbar. La duramadre expandida puede presionar los nervios al salir de la médula espinal. Esto puede causar dolor de espalda y también debilidad y entumecimiento en las piernas. También puede causar dolores de cabeza.
Continúa leyendo abajo
¿Qué causa el síndrome de Marfan?
Volver al contenidoEl síndrome de Marfan es causado por un cambio (mutación) en el material genético de uno de tus cromosomas (cromosoma número 15). El gen que se ve afectado es responsable de producir una proteína especial llamada fibrilina. El gen se llama gen de la fibrilina 1 (FBN1). La fibrilina es una parte importante del tejido conectivo en el cuerpo. Ayuda a fortalecer el tejido conectivo y hacerlo elástico. Si no se produce suficiente fibrilina, esto puede llevar a un tejido conectivo 'flojo' y débil. La fibrilina se encuentra en el tejido conectivo en muchas partes de tu cuerpo, incluyendo tus ojos, tus vasos sanguíneos, tu corazón y tus huesos.
Tres de cada cuatro personas que tienen el síndrome de Marfan tienen un padre que también tiene el síndrome de Marfan. Puedes heredar el síndrome de Marfan de solo uno de tus padres. Se conoce como un trastorno autosómico dominante, lo que significa que solo uno de tus padres necesita tener el gen defectuoso para transmitírtelo.
Una de cada cuatro personas que tienen el síndrome de Marfan lo tiene debido a un nuevo cambio en el material genético ('nueva mutación'). Ninguno de sus padres tiene la condición. Por alguna razón, el cambio en el gen de la fibrilina ocurrió por primera vez en el óvulo o esperma de uno de sus padres.
¿Cómo se diagnostica el síndrome de Marfan?
Volver al contenidoEl diagnóstico del síndrome de Marfan incluye revisar tu historial familiar, a veces realizar algunas pruebas genéticas, y también observar si diferentes partes de tu cuerpo presentan alguno de los problemas típicos.
Su médico generalmente comenzará haciéndole preguntas sobre su familia y preguntando sobre cualquier síntoma que pueda tener que sugiera el síndrome de Marfan. Luego, pueden examinarlo para ver si tiene alguna de las características típicas descritas anteriormente.
El síndrome de Marfan a veces puede ser diagnosticado en el útero antes del nacimiento, o poco después del nacimiento. Para la mayoría de las personas, el síndrome de Marfan no se diagnostica hasta más tarde en la infancia o en la adultez. Esto se debe a que puede tomar tiempo para que las características y problemas típicos del síndrome de Marfan se hagan evidentes.
Su médico de familia generalmente lo derivará a un especialista si sospecha que puede tener el síndrome de Marfan. Esto permitirá realizar más pruebas. Estas pruebas pueden incluir:
Radiografía de tórax to look for signs of a widened main blood artery in your body (the aorta).
Una ecografía del corazón (ecocardiograma, o eco) to look for heart problems and any widening of the aorta.
Tomografía computarizada (TC) o escaneo de resonancia magnética (IRM) of the chest to look at the aorta, and also of the spine to look for any signs that the membrane surrounding your brain and spinal cord (called the dura) may have become weak and ballooned outwards (known as dural ectasia).
Registro cardíaco de 24 horas (electrocardiograma, o ECG) to look for problems with the rhythm of your heartbeat.
Un examen de tus ojos para buscar una dislocación del cristalino o problemas con el revestimiento en la parte posterior de tu ojo (retina).
Radiografías de su cadera y pelvis, que pueden mostrar un problema con su cadera.
Pruebas genéticas - there is no single genetic test that can diagnose Marfan syndrome. This is because not all people with the abnormal gene have Marfan syndrome and in some people with Marfan syndrome the abnormal gene can't be found. Genetic testing is usually done to look for gene mutations. As time goes on, more different genetic mutations are being found.
Esta imagen muestra una prueba informal para el síndrome de Marfan, llamada el 'signo de la muñeca': si la persona con síndrome de Marfan agarra su muñeca con la mano, su dedo meñique se superpone a su pulgar. Esto ocurre en aproximadamente 6 de cada 10 personas con síndrome de Marfan. Es posible pero raro tener este signo sin el síndrome de Marfan.
Signo de la muñeca en el síndrome de Marfan
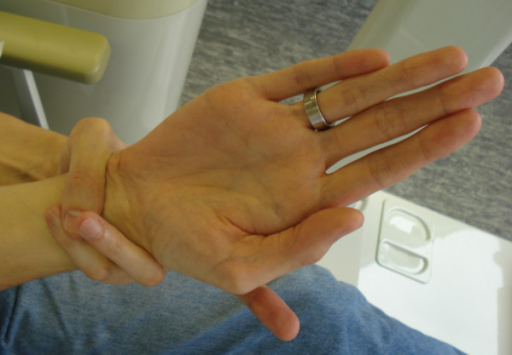
© Staufenbiel I, Hauschild C, Kahl-Nieke B, Vahle-Hinz E, von Kodolitsch Y, Berner M, Bauss O, Geurtsen W, Rahman A, CC BY-SA 2.0, a través de Wikimedia Commons
Doctors may use the 'Ghent criteria' to help diagnose Marfan syndrome. These include a list of some of the typical features of Marfan syndrome. There needs to be a certain number of features present plus either a relative who has Marfan syndrome or an abnormal genetic test to make a diagnosis of Marfan syndrome.
Continúa leyendo abajo
¿Cuál es el tratamiento para el síndrome de Marfan?
Volver al contenidoNo hay cura para el síndrome de Marfan. Sin embargo, hay tratamientos disponibles para ayudar con los problemas que causa el síndrome de Marfan. Debido a que el síndrome de Marfan puede afectar diferentes partes de tu cuerpo, es posible que te atiendan diferentes especialistas. Estos especialistas pueden incluir un especialista en corazón (cardiólogo), un especialista en ojos (oftalmólogo), un especialista en huesos y articulaciones (cirujano ortopédico) y un especialista en problemas genéticos (genetista).
¿Cuándo se necesita tratamiento de emergencia?
Las personas con síndrome de Marfan tienen un mayor riesgo de ciertas condiciones que requieren tratamiento hospitalario de emergencia. Por favor, busque ayuda de inmediato si cree que puede tener alguna de las siguientes:
Disección aórtica: puede causar un dolor torácico repentino, intenso, agudo o desgarrador y dificultad para respirar. El dolor puede irradiarse hacia la espalda o el abdomen. A menudo, el dolor se extiende al cuello o baja por la espalda. Otros síntomas pueden incluir pérdida de conciencia.
Desprendimiento de retina: los síntomas pueden incluir luces intermitentes, manchas flotantes y pérdida de visión. Puede notar sombras en su visión periférica, visión nublada o pérdida de visión como si una cortina cubriera su ojo.
Colapso pulmonar repentino (neumotórax espontáneo): el síntoma típico es la aparición repentina de un dolor agudo y punzante en un lado del pecho. El dolor suele empeorar al inhalar. También puede experimentar dificultad para respirar.
Tratamiento para problemas del corazón y vasos sanguíneos
Si tienes el síndrome de Marfan, necesitarás chequeos regulares para buscar y monitorear cualquier problema del corazón y los vasos sanguíneos. Por ejemplo, es posible que te realicen ecocardiografías regulares (escaneos del corazón) para verificar el ensanchamiento de tu arteria principal (aorta).
There are medicines which may help to slow down any widening of your aorta. Betabloqueantes are often prescribed for this reason. Other medicines used include antagonistas del calcio, inhibidores de la ECA y bloqueadores de los receptores de angiotensina.
Sometimes heart surgery is needed to repair or replace part of the main artery if it becomes too wide. This is usually advised once the size of the aorta reaches 5cm. This is because there is a risk that the artery may tear or burst suddenly. During surgery, the dilated part of the artery is replaced using a graft. Sometimes the valve between the heart and main blood artery (the aortic valve) is replaced as well. In some people, emergency surgery is needed if the main artery tears or bursts, but the aim of treatment is to operate on the bulging aorta before this. The outcomes from planned surgery are much better than the outcomes from emergency surgery.
La cirugía cardíaca también puede ser necesaria para problemas de la válvula mitral.
Tratamiento para problemas del esqueleto
Algunos de los problemas con tus huesos que pueden ocurrir con el síndrome de Marfan pueden requerir tratamiento. La fisioterapia se utiliza a menudo para el dolor de cadera, dolor de espalda o problemas al caminar.
An abnormal curve of your spine (scoliosis) may be treated with a back brace. The idea is that the brace will stop the abnormal curve from becoming any worse. A brace does not cure the abnormal curve of your spine. It may be particularly useful for children who are still growing. If severe then the abnormal curve of your spine may need surgery. Consulte el folleto separado titulado Escoliosis y cifosis (Curvatura de la columna vertebral) para más detalles.
Si tu esternón está hundido hacia adentro (pectus excavatum), tus pulmones y respiración pueden verse afectados y podrías necesitar cirugía para corregirlo.
Si tu esternón está empujado hacia afuera (pectus carinatum), esto generalmente no causa ningún problema. Algunas personas con el esternón empujado hacia afuera eligen someterse a cirugía correctiva por razones estéticas.
A veces, también puede ser necesaria la cirugía para corregir problemas de cadera.
Tratamiento para problemas oculares
See also the separate leaflet Anatomía del ojo.
Serás atendido regularmente por un especialista en ojos si tienes el síndrome de Marfan.
Es posible que se necesite tratamiento para problemas oculares. Por ejemplo, se pueden usar láseres para reparar un problema con el revestimiento en la parte posterior de su ojo (retina desprendida).
Si el cristalino de su ojo se desplaza a una posición anormal (cristalino dislocado), puede ser necesaria una cirugía para extraer el cristalino del ojo.
If you develop clouding of the lens within your eye (a cataract), you may need the lens in your eye to be removed and an artificial lens put in its place. Consulte el folleto separado llamado Cataratas para más detalles.
Si desarrollas un aumento de presión dentro de tu ojo (glaucoma), los tratamientos pueden incluir varios colirios, medicamentos, tratamiento con láser y cirugía.
Glasses or contact lenses may be needed if you have short sight (myopia).
Cambios en el estilo de vida
La mayoría de las personas con síndrome de Marfan pueden llevar una vida relativamente normal. Sin embargo, el levantamiento de pesas y algunos deportes competitivos y de contacto pueden no ser adecuados si tienes síndrome de Marfan.
También se le puede aconsejar evitar el buceo y la escalada. Esto se debe a la debilidad en la arteria principal (aorta) y el ojo, y a cualquier problema esquelético que pueda tener. Su especialista le aconsejará si debe evitar alguna actividad específica.
Se recomiendan muchas actividades y deportes como caminar, jugar al golf y pescar si tienes el síndrome de Marfan.
Otros tratamientos
Ser diagnosticado con el síndrome de Marfan puede ser algo difícil de afrontar para algunas personas. Para algunas personas, la altura y la apariencia pueden causar preocupaciones y afectar la confianza y la autoestima. La posibilidad de que se necesite una cirugía mayor, por ejemplo, una cirugía cardíaca, también puede ser algo difícil de manejar.
Puede resultarle útil unirse a un grupo de apoyo para que pueda ponerse en contacto con otras personas que tienen el síndrome de Marfan. También puede encontrar útil recibir asesoramiento individual tanto para usted como para los miembros de su familia.
There may also be some other specialist treatments available for Marfan syndrome. One example is hormone treatment to bring on puberty early in children so that their adult height can be reduced. Your specialist will be able to advise you about the latest treatments.
¿Puedo transmitir el síndrome de Marfan a mis hijos?
Volver al contenidoSí - si tienes el síndrome de Marfan, por cada hijo que tengas, hay una probabilidad del 50:50 de que también tenga el síndrome de Marfan. Esto también significa que hay una probabilidad del 50:50 de que cada hijo no tenga el síndrome de Marfan.
Herencia autosómica dominante
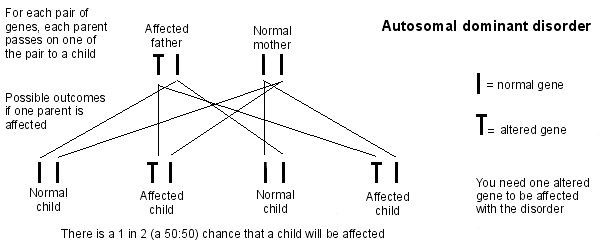
Testing for Marfan syndrome in your baby when you are pregnant can be done using muestreo de vellosidades coriónicas o amniocentesis. Puede ser capaz de mostrar si su bebé tiene el gen defectuoso. Sin embargo, debe recordar que el síndrome de Marfan es una enfermedad variable. Afecta a diferentes personas de diferentes maneras. Así que un hijo que tenga puede verse afectado menos severamente, o más severamente, que usted.
Si eres una mujer con síndrome de Marfan y quedas embarazada, el embarazo puede ejercer una mayor presión sobre tu corazón. Esto puede aumentar la probabilidad de que la arteria principal se rompa (ruptura aórtica). Necesitarás un seguimiento regular con un especialista si tienes síndrome de Marfan y quedas embarazada.
Si tú (o tu pareja) tienes el síndrome de Marfan y deseas tener un hijo, es posible que quieras buscar el consejo de un especialista en problemas genéticos (un genetista) antes de quedar embarazada.
¿Cuál es la perspectiva?
Volver al contenidoEl pronóstico para las personas con síndrome de Marfan es muy variable. Algunas personas con el síndrome solo tienen unos pocos problemas que no causan muchas dificultades. Otras personas con el síndrome pueden verse gravemente afectadas.
Si no se tratan los problemas cardíacos, pueden llevar a la muerte en algunos casos. Esta es la razón principal por la que es importante que el síndrome de Marfan se reconozca temprano para que puedas tener un monitoreo continuo, especialmente de cualquier problema cardíaco. El reconocimiento temprano del síndrome de Marfan también significará que se puede llevar a cabo un tratamiento para evitar complicaciones graves. Antes de los nuevos medicamentos y las cirugías tempranas para problemas cardíacos, la esperanza de vida promedio con el síndrome de Marfan era de 32 años (en 1972); la esperanza de vida promedio ahora es similar a la de alguien sin el síndrome de Marfan.
Las principales causas de muerte prematura en el síndrome de Marfan ahora son en personas que no han sido diagnosticadas o que son diagnosticadas muy tarde y cuando ya ha ocurrido demasiado daño al corazón.
Patient picks for Condiciones genéticas

Salud infantil
Rasgo de células falciformes
Si heredas un gen de uno de tus padres, llamado gen de células falciformes, tienes una condición llamada rasgo de células falciformes. Si heredas un gen de células falciformes de ambos padres, obtienes una doble dosis del gen de células falciformes. Esto causa una condición llamada enfermedad de células falciformes (ECF).
by Dr Sarah Jarvis

Salud infantil
Trastornos del almacenamiento de glucógeno
Los trastornos del almacenamiento de glucógeno son un grupo de enfermedades hereditarias. Resultan de un problema con una de las proteínas (conocidas como enzimas) involucradas en la conversión de glucosa en glucógeno, o en la descomposición del glucógeno de nuevo en glucosa. Afectan principalmente al hígado y a los músculos. La mayoría se diagnostican en la infancia. Los síntomas incluyen debilidad, fatiga y niveles bajos de azúcar en la sangre. Las nuevas opciones de tratamiento ofrecen esperanza para un pronóstico mejorado. La mayoría de las personas con un trastorno del almacenamiento de glucógeno responden bien al tratamiento. Sin embargo, la tipo II (enfermedad de Pompe infantil) puede ser difícil de tratar y afectar la esperanza de vida.
por el Dr. Roger Henderson, MBBS
Preguntas frecuentes
What is connective tissue and why is it important in Marfan syndrome?
Connective tissue is a vital part of your body that provides structure, strength, and protection to your organs and other tissues. It also helps transport fat and nutrients and assists in repairing damaged tissues. In Marfan syndrome, this connective tissue becomes weak and 'floppy' due to a genetic problem with a protein called fibrillin, which can lead to issues in various parts of the body.
Is Marfan syndrome always inherited from a parent?
No, not always. While it is true that about three out of four people with Marfan syndrome inherit it from a parent who also has the condition (it's an autosomal dominant disorder), for about one in four people, the condition arises from a 'de novo' mutation. This means the genetic change happened for the first time in the egg or sperm of one of their parents, so neither parent has the condition.
Can Marfan syndrome symptoms appear differently in various individuals?
Yes, Marfan syndrome can affect people very differently. Not all parts of the body are affected in everyone, and some individuals may have only a few mild symptoms, while others are more severely affected. The symptoms also tend to worsen as a person gets older.
What is the wrist sign for Marfan syndrome and is it definitive for diagnosis?
The wrist sign is an informal test where if a person with Marfan syndrome grips their wrist with their hand, their little finger overlaps their thumb. This occurs in about 60% of people with the condition. However, it's not a definitive diagnostic tool; it's possible, though rare, to have this sign without having Marfan syndrome. Diagnosis usually involves a combination of family history, physical examination, and various medical tests.
Are there any sports or activities I should avoid if I have Marfan syndrome?
Yes, if you have Marfan syndrome, you may be advised to avoid weightlifting and some competitive and contact sports, as well as scuba diving and climbing. This is due to potential weakness in your main blood artery (aorta), eye issues, and skeletal problems. Your specialist will be able to advise you on which specific activities you should avoid.
What is the life expectancy for someone with Marfan syndrome today?
Thanks to new medications and earlier surgery for heart problems, the average life expectancy for someone with Marfan syndrome is now similar to that of someone without the condition. Historically, in 1972, the average life expectancy was much lower, around 32 years. Early diagnosis and ongoing monitoring, especially of heart problems, are crucial to achieving this improved outlook.
Lecturas adicionales y referencias
- Síndrome de Marfan, MFS; Herencia Mendeliana en Línea en el Hombre (OMIM)
- Staufenbiel I, Hauschild C, Kahl-Nieke B, et al; Condiciones periodontales en pacientes con síndrome de Marfan - un estudio de caso control multicéntrico. BMC Oral Health. 28 de octubre de 2013;13:59. doi: 10.1186/1472-6831-13-59.
- Salik I, Rawla P; Marfan Syndrome.
- Revisión del síndrome de Marfan: De la genética a la práctica clínica; Science Direct
Continúa leyendo abajo
About the authorView full bio

Dra. Philippa Vincent, MRCGP
Médico General, Autor Médico
MB BS, Bsc, MRCGP (2000), DCH, DFSRH, DRCOG
Dra Philippa Vincent is an NHS GP working in North London.
About the reviewerView full bio

Dr Rosalyn Adleman, MRCGP
MRCGP
Dr Rosalyn Adleman, is an NHS GP working in north London.
Historial del artículo
La información en esta página está escrita y revisada por pares por clínicos calificados.
Próxima revisión: 2 Mar 2028
3 Mar 2025 | Última versión

Pregunta, comparte, conecta.
Navega por discusiones, haz preguntas y comparte experiencias en cientos de temas de salud.

¿Te sientes mal?
Evalúa tus síntomas en línea de forma gratuita
Suscríbete al boletín de Patient
Tu dosis semanal de consejos de salud claros y confiables, escritos para ayudarte a sentirte informado, seguro y en control.
By subscribing you accept our Política de Privacidad. Puedes darte de baja en cualquier momento. Nunca vendemos tus datos.